
Eosinophilic Fasciitis (Shulman’s Syndrome) is a disease that has been diagnosed fewer than 300 times in the past 35 years – that’s 9 cases per year in the US! Eosinophils, a type of white blood cell, permeate the muscle tissue (fascia), which becomes hard, swollen and inflexible. This can occur in the arms, legs, shoulders, hands, feet, face and trunk. Diagnosis is made with an open muscle biopsy, and treatment is typically corticosteroids and other immune-supressing medications. Blood disorders and blood cancers are common complications. With no known cause and so few documented cases, EF is challenging to treat.
Learn More about EF:
I think the initials EF are especially fitting for such an effed-up disease 🙂

If you know me, you know I’ve always strived for uniqueness/out-of-the-boxness/one-of-a-kindness. Uh, yah, I was kinda focused more along the lines of creative projects – not my health.
After spending beaucoup d’hours at oncologists’ offices and the Hoag Hospital Cancer Center, I am very thankful that I have EF, and not one of the much more brutal diseases that most of my fellow patients have. It could be SO much worse!!! UPDATE: It actually has become worse, as I have developed a life-threatening complication called Severe Aplastic Anemia, which is basically bone marrow failure. My brawl just got a little more serious, but I will battle and kick AA’s sorry ass 🙂
Eosinophilic Fasciitis is sometimes followed by various blood disorders, such as Leukemia, Lymphoma or Aplastic Anemia. In my case, my blood tests showed that I was losing cells in all three major parts of my blood: white cells, red cells, and platelets. After my second bone marrow biopsy, I was diagnosed with Severe Aplastic Anemia, which is another very rare condition, occurring in 1 or 2 people per million. My doctors at the time tried to raise my counts by using different drugs: Solumedrol (3 days of IV form of super concentrated Prednisone), IVIG (5 days of Intravenous Immune Globulin), Neupogen (injections that stimulate white blood cell production) and high doses of Prednisone and Cyclosporine. While we were waiting for the counts to rise, I received several transfusions each week of red blood cells and platelets to keep me alive.
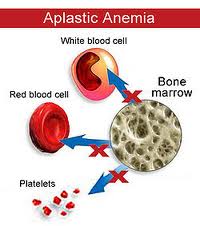 After realizing that this was not the best path to deal with my Aplastic Anemia, we started seeing specialists at City of Hope. There, I was hospitalized while being transfused with ATG (4 days of IV Anti-thymocyte Globulin – a form of chemo derived from horse antibodies). I picked up a fungal infection in my lung, which is very dangerous with high doses of immune suppressants and virtually no white blood cells to fight it, so I stayed for 45 days. I did not respond to the ATG, so my team of doctors decided I needed a bone marrow transplant, the last resort. About a month later, I was re-admitted to the COH Helford Hospital, and was given high doses of chemo for 4 days, which was intended to wipe out my diseased bone marrow and my immune system in preparation for a friendly welcome to the new marrow. My brother was a perfect match, and on December 14, 2012, I received a quart of his bone marrow during my transplant. We started seeing improvements in my counts about 3 weeks post transplant, and they have continued to rise. I was released after a second 45 days, when my docs were confident that my counts were on an upward path and all my IV meds were being tolerated in pill form. After a fourth bone marrow biopsy, it was determined that I am presently 99.75% my brother on a cellular level, which is allowing me to make my own healthy white cells, red cells and platelets. My doc said I could go on a crime spree and pin it on him 🙂 So I have that going for me… I’m just praying I don’t suddenly sprout hairy knuckles. There is a long road of recovery ahead of me, but it appears my Aplastic Anemia is getting slayed 🙂
After realizing that this was not the best path to deal with my Aplastic Anemia, we started seeing specialists at City of Hope. There, I was hospitalized while being transfused with ATG (4 days of IV Anti-thymocyte Globulin – a form of chemo derived from horse antibodies). I picked up a fungal infection in my lung, which is very dangerous with high doses of immune suppressants and virtually no white blood cells to fight it, so I stayed for 45 days. I did not respond to the ATG, so my team of doctors decided I needed a bone marrow transplant, the last resort. About a month later, I was re-admitted to the COH Helford Hospital, and was given high doses of chemo for 4 days, which was intended to wipe out my diseased bone marrow and my immune system in preparation for a friendly welcome to the new marrow. My brother was a perfect match, and on December 14, 2012, I received a quart of his bone marrow during my transplant. We started seeing improvements in my counts about 3 weeks post transplant, and they have continued to rise. I was released after a second 45 days, when my docs were confident that my counts were on an upward path and all my IV meds were being tolerated in pill form. After a fourth bone marrow biopsy, it was determined that I am presently 99.75% my brother on a cellular level, which is allowing me to make my own healthy white cells, red cells and platelets. My doc said I could go on a crime spree and pin it on him 🙂 So I have that going for me… I’m just praying I don’t suddenly sprout hairy knuckles. There is a long road of recovery ahead of me, but it appears my Aplastic Anemia is getting slayed 🙂
Learn more about AA:

Is that kinda like ENFU having EF?? BTW… I know where this picture was taken and I was there with you. Can’t believe that’s been two years ago…where has time gone. Love the nails, yes creative, different and so you! That’s why we love YOU!
LikeLike
Hi!
I too am afflicted by the syndrome. I live in Ontario Canada. I was diagnosed in January 2011 and have had some success with Prednisone. I was told this rare syndrome is present in about 1 in 200,000 person.
When were you diagnosed and have you had some flare ups? I am presently having mild flare ups in a left arm and the doc wants me to wait to go back on Prednisone again.
LikeLike
Hi Rachel – Sorry to hear that you are having flare ups. I have a love/hate relationship with Prednisone. It works when you need it, but the side effects are tough to deal with, right? I hope you can get on a low dose and it clears up your EF soon, so you can get off it asap 🙂 I was diagnosed in the summer of 2012, and I was given Prednisone and Cyclosporin to clear it up. I couldn’t really bend my arms or legs, open my mouth very wide, raise my arms or sit down without pain. My EF was completely cured with these meds, but it went into Severe Aplastic Anemia, which required a bone marrow transplant. I am now in remission 🙂 Thanks for reading my blog, Rachel, and I wish you all the best in your battle with EF!
LikeLike
I hope you are are doing well Joselyn, I miss your posts. These are very uncertain times, just wanted to send you and your family lots of love. I also got diagnosed with EF, but doing a whole lot better.
LikeLike
Hi Louise – Thanks for the kind message 🙂 Yes, these times are uncertain, but I’m trying to find silver linings 🙂 I’m sorry about your diagnosis, but so glad to hear that you are doing better!!!
LikeLike